



「禁忌を含む注意事項等情報」等については電子添文をご参照ください。
非盲検継続投与試験(OLE)
- 1) Judge DP, et al. Circulation. 2024; Online ahead of print.
- [COI:著者のなかにはAlexion Pharmaceuticals, Inc. 及び AstraZeneca plc., BridgeBio Pharma, Inc. よりコンサルティング料等を受領している者が含まれる]
|
ビヨントラ®の有効性と安全性
ー非盲検継続投与(OLE)試験ー
1. 試験概要
| 目的 | 症候性のATTR-CM患者を対象にビヨントラ®の長期継続投与における有効性及び安全性を評価する。 |
|---|---|
| 対象 |
非盲検継続投与(Open Label Extension:OLE)適格基準*を満たした症候性の野生型又は変異型ATTR-CM患者 389例 |
| 方法 |

|
| リミテーション | OLEは非盲検であり、真の対照群を欠くため長期安全性データの解釈に避けられない不確実性をもたらす。ATTRibute-CM試験参加者のうち、OLE非参加者の存在は治療効果の推定力を低下させ、また治療効果はOLE参加者の非ランダム性に影響された可能性がある。ATTRibute-CM試験のプラセボ群は、治療効果を得たビヨントラ群と比較し、より大きな病勢進行を経験している。そのため、OLE試験参加者の病勢進行に関するベースライン特性には偏りがあり、治療の推定効果に影響を与えた可能性がある。 |
| 主要評価項目 |
|
| 副次評価項目 |
|
| その他の評価項目 |
|
| 解析計画 |
解析対象集団
解析方法
|
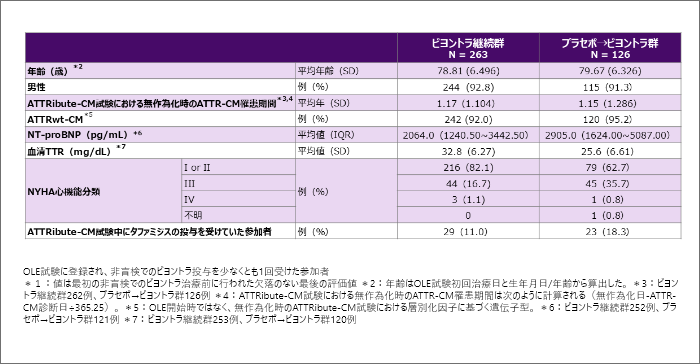
患者背景*1
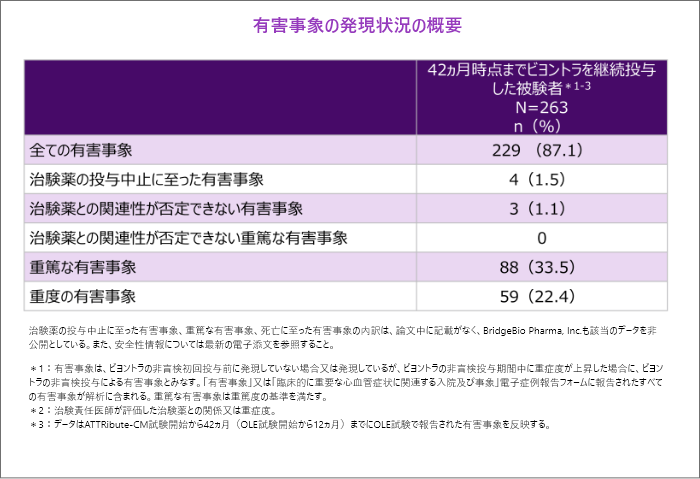
2. 安全性[主要評価項目]42ヵ月
- 42ヵ月時点で有害事象は87.1%に認められ、そのうち治験薬との関連性が否定できない有害事象は1.1%でした。
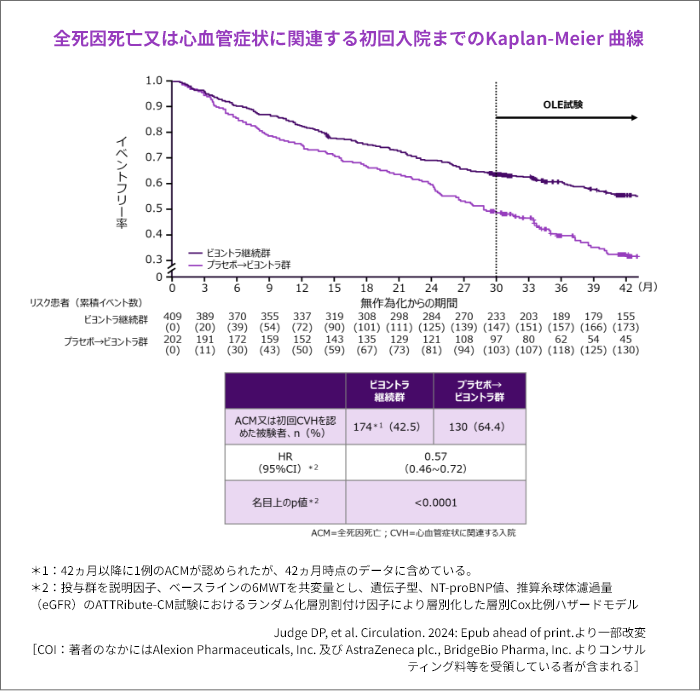
3. 有効性 42ヵ月
全死因死亡又は心血管症状に関連する初回入院の累積頻度の複合エンドポイント[副次評価項目]
- ビヨントラ継続群はプラセボ→ビヨントラ群と比較し、42ヵ月時点までで2構成要素のイベント発現リスクが43%低下しました。
全死因死亡[副次評価項目]42ヵ月
- 36ヵ月時点まででビヨントラ継続群とプラセボ→ビヨントラ群に有意差が認められました。
- ビヨントラ継続群はプラセボ→ビヨントラ群と比較し、36ヵ月時点、42ヵ月時点までの各期間で全死因死亡のリスクがともに36%低下しました。
![全死因死亡のKaplan-Meier曲線Judge DP, et al. Circulation. 2024: Epub ahead of print.より一部改変 [COI:著者のなかにはAlexion Pharmaceuticals, Inc. 及びAstraZeneca plc., BridgeBio Pharma, Inc. よりコンサルティング料等を受領している者が含まれる]](/-/media/beyonttra_jp/attributecm_ole/group4000_03_4.png?rev=-1&la=ja-JP&h=734&w=700&hash=E92A25BC18B38D20F5B05C5B0D2A65C5)
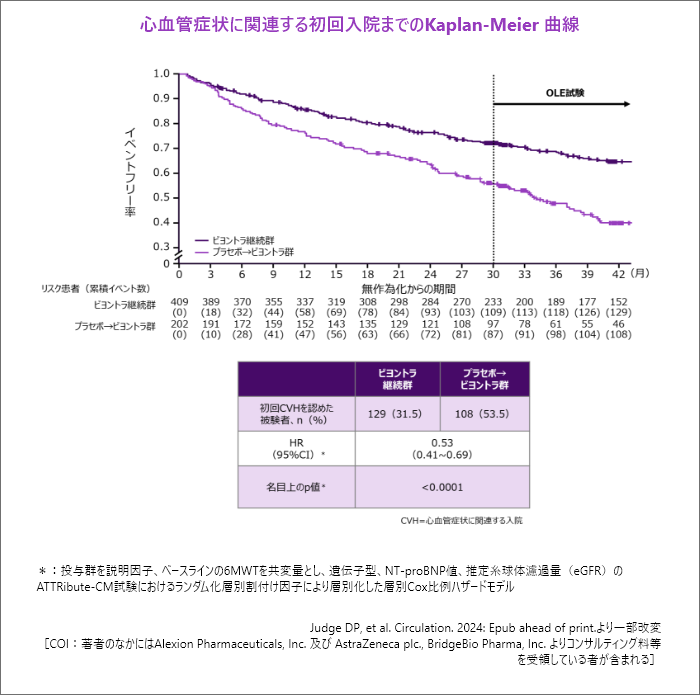
心血管症状に関連する初回入院までの期間[副次評価項目]42ヵ月
- ビヨントラ継続群はプラセボ→ビヨントラ群と比較し、42ヵ月時点までで心血管症状に関連する入院リスクが47%低下しました。
NT-proBNPのベースラインからの変化量[その他の評価項目] 42ヵ月
- 42ヵ月時点のNT-proBNPのベースラインからの倍数変化の幾何平均は、ビヨントラ継続群で1.10、プラセボ→ビヨントラ群で2.29でした。
- ATTRibute-CM試験で確認された両群の差は、OLE試験において下図の通りに推移しました。
![NT-proBNPのベースラインから42ヵ月時点までの経時的な変化量Judge DP, et al. Circulation. 2024: Epub ahead of print.より一部改変[COI:著者のなかにはAlexion Pharmaceuticals, Inc. 及びAstraZeneca plc., BridgeBio Pharma, Inc. よりコンサルティング料等を受領している者が含まれる]](/-/media/beyonttra_jp/attributecm_ole/group4000_03_6.png?rev=-1&la=ja-JP&h=431&w=700&hash=3C401DD4D1D858818682ECD96E263E46)
プラセボ+タファミジス群がビヨントラに切り替えた際の血清TTRレベルの変化量[その他の評価項目] 36ヵ月
- プラセボ+タファミジス→ビヨントラ群における血清TTRレベルの平均変化量は、OLE試験開始時(ATTRibute-CM試験30ヵ月時点)と比較して上昇し、1ヵ月時点で3.0mg/dL、6ヵ月時点で3.4mg/dLでした2)。
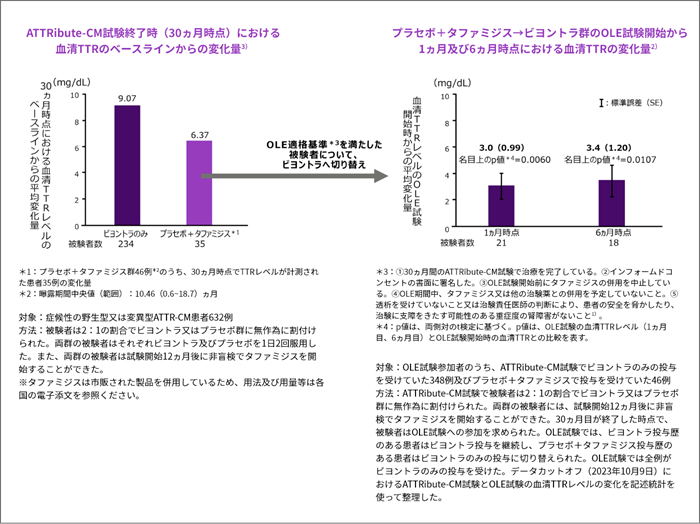
- 2) Maurer M, et al. European Heart Journal. 2024; 45(Suppl 1) : ehae666.2088.
[COI: 本研究はBridgeBio Pharma, Inc. の資金提供により行われた] - 3) 社内資料:臨床的有効性の概要(承認時評価資料)
電子添文は、こちらよりご覧いただけます。


